
When should you bring a Quantitative Clinical Pharmacologist into your program?
Earlier than you think.
Why bring an expert early on
The non-clinical phase is the foundation a drug programme is built on. Compounds are being optimised and characterised in terms of ADME, in vitro assays are run to identify DDI and cardiovascular liabilities, and for compounds of interest metabolites may be profiled. These are the building blocks that determine whether a compound is worth taking forward. But there is one question that sits above all of them.
What is the efficacious human dose?
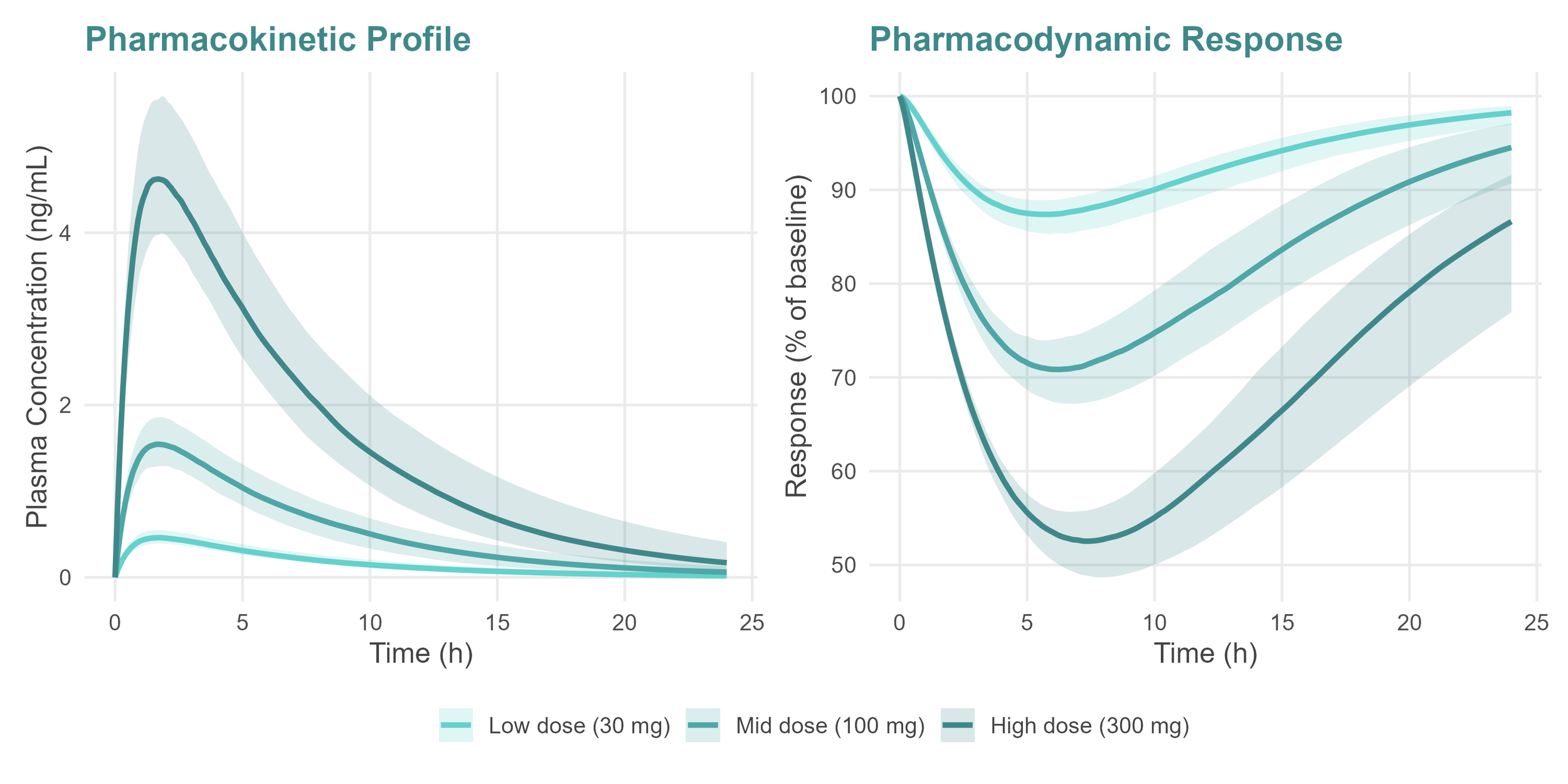
This is the central question that is not only important to assess whether a compound is likely to have the correct profile to be considered a clinical candidate, but it also defines a cascade of activities across other critical areas: formulation development, toxicology study design, clinical dose escalation strategy, and the overall development plan. All of these flow from a single number, or more precisely, from a predicted exposure target and the regimen needed to achieve it. Get it wrong and the consequences compound at every subsequent stage. Get it right early, and every decision that follows is built on solid ground.
This is precisely the stage where quantitative analyses, modelling and simulation and clinical pharmacology experience add the most value. Not as a reactive exercise when questions are already being asked by regulators, but embedded throughout the entire non-clinical phase, from early study design to candidate selection, shaping the questions each experiment is designed to answer.
What does a human dose prediction give you
Human dose predictions are often derived using fit-for-purpose PKPD models combined with translational strategies, allowing a prediction grounded in the biology of the compound rather than convention or assumption [1, 2]. But the value goes beyond a single number. It reflects a deep understanding of the mechanism of action of the compound in question. Once the desired exposure eliciting a pharmacological effect is understood and integrated with the PK profile of the compound, discussions around dose and dosing regimen feed directly into the clinical development strategy.
This matters more than ever in the current regulatory environment. With Project Optimus now firmly in place for oncology programmes, and the MIDD framework laid out by the FDA [1], sponsors are expected to explore a range of doses predicted to be efficacious, or provide a clear rationale for dose selection, rather than simply identifying the maximum tolerated dose and working backwards. That requires a quantitative, evidence-based approach that can only come from robust non-clinical PKPD modelling [2].
Predicting a number should never be the end goal. The aim is to build a coherent, mechanistic understanding of the exposure-response relationship early enough that it can actually inform decisions, de-risking the programme and increasing the chances of success in the clinic. That said, the human dose prediction remains the most tangible outcome of the translational exercise from non-clinical data, and the one that will be communicated to and relied upon by every other function in the programme.
How dose prediction shapes the entire programme
A dose prediction is only as robust as the evidence and understanding behind it. When the non-clinical programme has not been designed with a quantitative understanding of exposure-response in mind, or when that relationship has not been sufficiently explored, the prediction rests on incomplete foundations. And incomplete foundations have a way of revealing themselves at the worst possible moment.
Take formulation as an example. If the compound ultimately requires higher exposures than initially predicted, reformulation may be necessary, requiring more time and effort in formulation development and a bridging bioequivalence study. In some cases, the revised dose cannot be stably formulated at all, creating a viability problem that no amount of modelling can retrospectively solve. Attempting to resolve these questions in the clinical stage is a risk no programme can afford, since chemistry production will already have been scaled to a certain formulation and changing course at that stage carries significant time and cost implications.
The toxicology understanding and perceived margins also carry risks without a solid prediction. The safety margins presented to regulators are calculated relative to the predicted human efficacious exposure. If that prediction rests on incomplete foundations, the therapeutic margins are built on false certainty. The coverage may appear adequate on paper while the reality in the clinic is very different, with implications for how far doses can be escalated and what regulators will permit.
Candidate selection is perhaps the most fundamental stage where modelling work is vital. Identifying the most promising compound means finding one that not only has the right ADME properties but can also be dosed on a regimen that is practical and viable. A compound with compelling biology but an unfavourable PK profile, or one that requires a regimen impossible to implement in practice, will not succeed regardless of its mechanism [3]. Understanding the predicted human dose and regimen early is therefore not just useful for planning. It is a critical input into the go/no-go decision on the compound itself.
Why experience and regulatory context matter
Building a robust dose prediction requires more than technical modelling skill. It requires the ability to integrate the full picture of non-clinical data across PK and PD, to recognise patterns that transfer from one compound to the next within the same chemical series, and to design each experiment with the next one already in mind. This iterative, cumulative way of working is not something that can be replicated by running a model in isolation. It develops over years of working inside drug development programmes, across functions, and across compounds.
Not everyone offering modelling support has had that exposure. Many have deep expertise in statistical modelling and computational methods, but have not spent years embedded in non-clinical teams, navigating the practical realities of study design: what time points are achievable given experimental constraints, how many samples can be taken during a certain time period, how to influence bioscientists to design the best and most informative PKPD studies. These are not trivial considerations. Without them, the non-clinical programme produces data, but not necessarily understanding.
There is also the question of regulatory context. Modelling does not exist in isolation from the broader development strategy. Understanding how a dose prediction fits into a regulatory submission, what regulators are likely to scrutinise, being able to argue the risk profile a mathematical model carries for the overall programme, are questions where a clinical pharmacology background, not just pharmacometrics, becomes indispensable [1, 2]. The most valuable contribution is not a model that reproduces the data, it is a quantitative framework that supports the right decision at the right time, and can be defended to a regulator.
This combination, deep non-clinical experience, cross-functional credibility, and regulatory fluency, is what BeMath brings to early-stage programmes.
References
Issa AM. Model-informed drug development in early-phase development: navigating complexity with quantitative clarity. Clinical Pharmacology in Drug Development. 2025; 14(10): 738–741. https://doi.org/10.1002/cpdd.1607
Sheng J, Zhang T. Advancing drug development with "fit-for-purpose" modeling informed approaches. Journal of Pharmacokinetics and Pharmacodynamics. 2025; 52: 52. https://doi.org/10.1007/s10928-025-09995-2
Kondic A, Bottino D, Harrold J, et al. Navigating between right, wrong, and relevant: the use of mathematical modeling in preclinical decision making. Frontiers in Pharmacology. 2022; 13: 860881. https://doi.org/10.3389/fphar.2022.860881