
FDA's new MABEL guidance isn't really about QSP. It's about something the agency left unaddressed for nearly two decades.
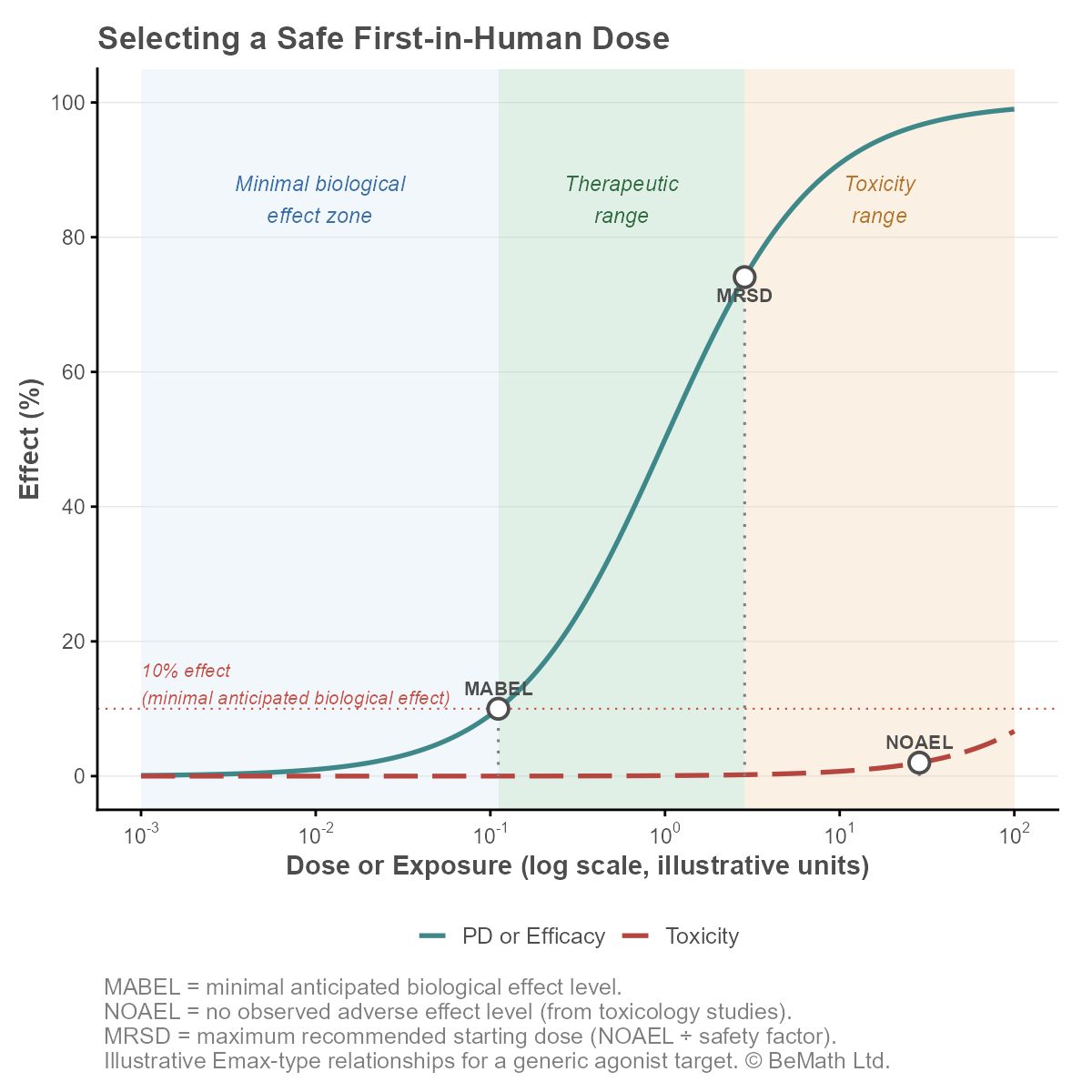
In June 2026, the Food and Drug Administration (FDA) published a draft guidance on using quantitative systems pharmacology (QSP) to determine the minimum anticipated biological effect level (MABEL) for first-in-human dose selection. On the surface, it reads as a methods document: how to build a model, what FDA expects to see in a pre-IND meeting, how to validate it.
Look a little closer, and this guidance is doing something more significant. It is the first time FDA has formally and structurally addressed MABEL at all, nearly two decades after a historical error caused serious harm to trial participants.
The gap nobody talks about
MABEL did not originate with a regulator. It was proposed in 2006 by a joint industry taskforce of the Association of the British Pharmaceutical Industry (ABPI) and the BioIndustry Association (BIA), formed in direct response to the TGN1412 trial, where six healthy volunteers in London received an anti-CD28 monoclonal antibody and, within an hour, experienced a severe systemic inflammatory response [1]. The UK Department of Health's Expert Scientific Group, chaired by Professor Gordon Duff, reviewed the incident and recommended the ABPI/BIA's MABEL concept over the conventional NOAEL-based approach [2].
TGN1412's starting dose was calculated as recommended by the regulatory standard of the time, but the animal model's toxicity findings did not translate to humans. Retrospective receptor occupancy calculations, performed independently by the ABPI/BIA taskforce and published separately by Horvath and Milton, suggest the dose administered resulted in receptor occupancy in excess of 90% [3]. A MABEL-based approach, targeting a lower receptor occupancy more typical for an agonist, would have pointed to a considerably more conservative starting dose.
The European Medicines Agency adopted MABEL into binding European Union guidance the following year. Under the current guideline, the starting dose should be related to MABEL, the pharmacologically active dose (PAD), or NOAEL depending on the level of uncertainty, and should generally be lower than the pharmacologically active dose unless a robust scientific rationale justifies otherwise [4].
FDA never built an equivalent binding requirement into general first-in-human guidance. MABEL surfaced in its documentation, but only in narrow, secondary contexts: a single paragraph in the ICH S9 Q&A confirming MABEL “could” be used for small molecules “if appropriate,” inside a guidance scoped specifically to anticancer pharmaceuticals [5]. Internal FDA staff analyses of CD3 bispecific constructs and checkpoint inhibitors examined MABEL too, but again within oncology, and as retrospective research rather than binding guidance [6].
The scope of the new June 2026 guidance is worth noting precisely because of this history. Unlike every prior FDA reference to MABEL, it is not confined to oncology or to immune-agonist mechanisms. The guidance applies to drugs that may, but does not necessarily, cause T-cell activation and cytokine release, and is framed for MABEL-based dose selection broadly [7]. For the first time, FDA is addressing MABEL as a general first-in-human concept, not a tool reserved for one therapeutic area.
That guidance gap did not mean MABEL was ignored in practice. In my own experience working in population pharmacokinetics and first-in-human dose selection, MABEL-informed thinking showed up in oncology submissions well before any FDA guidance required it, because sponsors, modellers, clinical pharmacologists and reviewers understood it was the right approach for high-risk biologics, even without being told to. The guidance gap was real, but the practice gap was smaller than the paper trail alone suggests.
Why the gap existed, and why “traditional MABEL” already existed too
The honest answer is not regulatory neglect. FDA's 2005 guidance on estimating the maximum safe starting dose, still cited in most first-in-human dose justifications today, discusses a pharmacology-based check, the pharmacologically active dose, or PAD, but is explicit about not going further: “Selection of a PAD depends upon many factors and differs markedly among pharmacological drug classes and clinical indications; therefore, selection of a PAD is beyond the scope of this guidance” [8].
The new guidance is more precise than it first appears on this point. The phrase “traditional MABEL estimates” appears once, defined only in passing as “those using in vitro pharmacology activity and binding studies conducted with the investigational product” [7]. The broader phrase “traditional methodologies” appears more than once, as shorthand for this same non-QSP approach, without ever being formally sourced. It does not need to be: this is, in substance, the methodology the European Medicines Agency has had in place since 2007, integrating receptor binding, occupancy and functional pharmacodynamic data into “a suitable modelling approach” that is explicitly platform-agnostic. Models linking PK/PD, and physiologically based pharmacokinetic (PBPK) models, are named as the relevant tools. QSP is not mentioned at all [4]. Still, FDA leaving the term undefined in its own guidance, rather than formally referencing the existing EMA framework or stating its own equivalent, is a documentation gap worth closing properly, not just one scientists can quietly fill by knowing where else to look.
This matters for how sponsors should read the new guidance: QSP is not the only, or even the default, route to a MABEL dose. For many compounds, especially those with a wide therapeutic index where the question is centered on pharmacodynamic effect than a narrow safety margin, a properly built PK/PD model using binding and occupancy data can answer the MABEL question without the cost and complexity of a full mechanistic QSP build. The guidance itself implicitly concedes this, telling sponsors to be cautious when a QSP-based dose differs substantially from “traditional MABEL estimates,” and to default to the more conservative figure regardless of which method produced it. A sponsor should reach for QSP when the simpler approach genuinely cannot capture what matters for that drug, target or disease, since MABEL itself does not require it.
What the new guidance actually does, then, is narrower in scope than a blanket QSP requirement. It gives FDA, for the first time, a detailed, prescriptive framework for the specific case where a sponsor needs or chooses a QSP model for MABEL: context of use, model risk assessment, parameterisation, verification, calibration, validation, sensitivity analysis. That structure was never written for "PAD" as a general 2005 concept. EMA had already shown a lighter-touch standardisation was achievable two years later, but FDA's 2005 guidance chose not to attempt it, leaving PAD as a comparison point rather than a defined methodology. It becomes possible once the model itself is fully specified and mechanistic, because the model has to do the justifying. EMA's lighter-touch, platform-agnostic guidance and FDA's new QSP-specific scaffolding are not competing definitions of MABEL. They answer two different questions: what MABEL requires, and what FDA expects to see when that reasoning is built into a QSP model specifically.
What sponsors are signing up for, if QSP is the right tool
None of what follows applies if a simpler MABEL estimate is sufficient for the compound in question. But for sponsors who do need QSP, the commitment is real.
The agency expects sponsors to bring a well-developed model design to the pre-IND or MIDD Paired Meeting stage, with model structure, key assumptions, critical parameters and data sources already specified, alongside clear plans for calibration, validation, sensitivity analysis and trial design [7]. The model development happens almost entirely on the sponsor's side, before FDA is meaningfully consulted, a real risk allocation decision on a development timeline measured in months rather than weeks.
The guidance is also asymmetric. If a QSP-derived dose is higher than a traditional MABEL estimate, sponsors must demonstrate the model's reliability, ideally through retrospective validation. If the QSP dose is lower, particularly for a novel target with no clinical precedent, sponsors are simply told to take the lower number. QSP can make a dose more conservative with relatively little friction; making it less conservative requires real evidentiary work.
Even after validation, the guidance does not let sponsors treat the model as the end of the safety question. It states explicitly that sensitivity and uncertainty analyses “can help, but cannot address, unanticipated biology,” and recommends staggered enrollment and intensive monitoring regardless of what the model predicts, with early clinical data expected to refine the model in real time [7]. The model informs the dose. It does not replace vigilance.
The bigger picture
None of this means QSP is mandatory, or that MABEL has become the only acceptable approach to first-in-human dosing. In current practice, NOAEL is rarely used alone; the standard approach compares a NOAEL-derived dose against a pharmacology-based estimate from a properly built PK/PD or PBPK model, and takes the more conservative of the two. For many compounds, that combination answers the MABEL question without QSP at all. But for high-risk biologics, novel targets, or anything where animal toxicology may not faithfully predict human pharmacology, this guidance gives the agency, for the first time, a structured path for getting a QSP-based MABEL right.
There is a line in the guidance's introduction that deserves more attention than it has had: a QSP-based approach “has the potential to reduce reliance on animal toxicology studies” [7]. It is mentioned once and left there. But it is worth sitting with. Animal toxicology studies are asked to predict how a drug will behave in human biology, and for TGN1412, dosed at Northwick Park Hospital in 2006, that is precisely what the cynomolgus monkey studies were unable to do. If a sufficiently detailed mechanistic model can credibly predict a safe, pharmacologically active human dose, the question is not just whether QSP can set a better starting dose. It is whether a well-validated QSP model could eventually do some of the work that animal toxicology is currently asked, and sometimes unable, to do, not replacing safety testing, but reducing how much of it has to come from a species whose biology was the actual problem nineteen years ago. This guidance does not attempt that conversation. But it is hard to read it and not wonder whether the agency already knows the question is coming.
References
[1] Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018–1028.
[2] Expert Scientific Group on Phase One Clinical Trials. Final report. London: Department of Health; 2006.
[3] Horvath CJ, Milton MN. The TeGenero incident and the Duff report conclusions: a series of unfortunate events or an avoidable event? Toxicol Pathol. 2009;37(3):372–383. https://doi.org/10.1177/0192623309332986
[4] European Medicines Agency. Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products. EMEA/CHMP/SWP/28367/07 Rev. 1. London: EMA; 2017. Section 7.2.
[5] U.S. Food and Drug Administration. S9 Nonclinical Evaluation for Anticancer Pharmaceuticals: Questions and Answers. Guidance for Industry. Silver Spring, MD: FDA; 2018. Q20.
[6] Saber H, Del Valle P, Ricks TK, Leighton JK. An FDA oncology analysis of immune activating products and first-in-human dose selection. Regul Toxicol Pharmacol. 2016;81:448–456.
[7] U.S. Food and Drug Administration. Quantitative Systems Pharmacology (QSP)-Based Dose Selection for Minimum Anticipated Biological Effect Level (MABEL) in First-in-Human (FIH) Trials. Draft Guidance for Industry. Silver Spring, MD: FDA; 2026.
[8] U.S. Food and Drug Administration. Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Guidance for Industry. Rockville, MD: FDA; 2005.